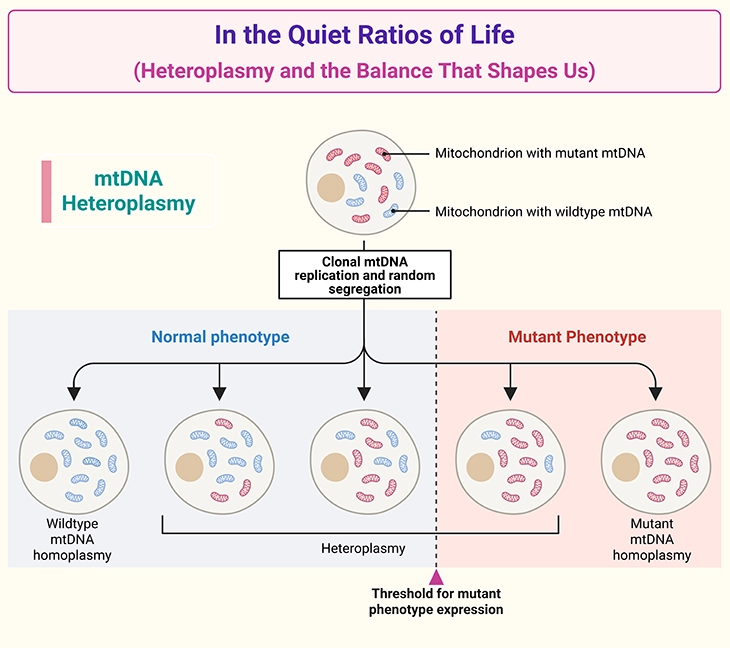
Figure 1. In the Quiet Ratios of Life: Heteroplasmy and the Balance That Shapes Us. Illustration of mitochondrial DNA (mtDNA) heteroplasmy and its impact on cellular function. Cells contain many mitochondria, each carrying multiple copies of mtDNA. Heteroplasmy refers to the coexistence of normal (wildtype) and mutant mtDNA within the same cell. The proportion of mutant mtDNA varies across cells, tissues, and individuals due to random segregation during egg formation and cell division. When the mutant load exceeds a critical threshold, mitochondrial ATP production becomes impaired, leading to tissuespecific symptoms. This threshold differs across organs, explaining the variable expression, severity, and clinical unpredictability of mitochondrial disorders — even within the same family.
Introduction
Mitochondria sit at the crossroads of life’s most fundamental processes, quietly powering every heartbeat, every neuronal spark, every moment of growth and repair. Though small, these organelles carry a legacy unlike any other in human biology: a second genome, inherited almost exclusively from the mother, passed down through generations like a whispered biological memory. This mitochondrial DNA (mtDNA) encodes essential components of the cell’s energy-producing machinery, and its integrity is vital for the health of organs with the highest metabolic demands — the brain, heart, muscle, liver, and kidneys.
At the center of mitochondrial function lies ATP (adenosine triphosphate), the universal energy currency that fuels every cellular task. The production of ATP depends on the coordinated activity of the mitochondrial respiratory chain, a fivecomplex system built from proteins encoded by both nuclear DNA and mtDNA. This dualgenome partnership is elegant but fragile. Variants in either genome — especially those affecting mtDNA — can disrupt energy production, leading to a spectrum of mitochondrial disorders whose severity depends on the proportion of working versus nonworking mitochondria within each cell.
This mixture, known as heteroplasmy, introduces a unique complexity to mitochondrial inheritance. Because mitochondria are passed through the egg, mtDNA follows a maternal inheritance pattern, yet the clinical expression of a variant is shaped by the threshold effect: symptoms emerge only when the proportion of nonfunctional mitochondria surpasses a critical level in enough tissues. This threshold varies across organs, reflecting their distinct energetic needs, and explains why mitochondrial conditions can vary dramatically within the same family — even between siblings (see Figure 1) [1-4].
Understanding these principles is essential not only for appreciating mitochondrial biology but also for recognizing its broader implications for human development. Emerging research reveals that mitochondrial dysfunction may contribute to neurodevelopmental differences, including autism spectrum disorder, where abnormalities in energy metabolism, oxidative stress, and mitochondrial enzyme activity have been reported in 30–50% of individuals in some studies. These findings underscore the importance of mitochondrial health as a foundation for brain development, resilience, and function [5-6].
In this series, we explore the intricate world of mitochondria — their genetics, their vulnerabilities, and their profound influence on health and disease. By weaving together molecular biology, developmental science, and clinical insight, we aim to illuminate how these tiny organelles shape the trajectory of human life, from conception to adulthood.
1.The Genetic Architecture of the Cell: Chromosomes, Genes, DNA, and Mitochondria
Our bodies are built from billions of cells, and within nearly every one of them lies a complete copy of our genetic “Book of Life.” This biological manuscript is written in the language of DNA, organized into structures called chromosomes, which reside in the nucleus—the cell’s central command center (see Figure 2).
Chromosomes and the Nuclear Genome
Each chromosome is composed of long, continuous strands of DNA, arranged as alternating stretches of genes—the proteincoding sequences—and noncoding regions that regulate when and how those genes are used. These chromosomes are present in the nucleus of almost all human cells, with one notable exception: red blood cells (RBCs). Mature RBCs eject their nucleus during development, allowing more room for hemoglobin and thus do not contain chromosomes or nuclear DNA.
Mitochondria: The Second Genome
Beyond the nucleus, cells contain another, much smaller repository of genetic information housed within mitochondria—tiny, energyproducing organelles scattered throughout the cytoplasm. Unlike the linear chromosomes of the nucleus, mitochondrial DNA (mtDNA) is arranged as a single circular molecule, reminiscent of bacterial genomes, reflecting mitochondria’s ancient evolutionary origins (see Figure 2).
Although mtDNA is compact—only 16,569 base pairs in humans—it encodes 37 genes, including 13 essential proteins of the oxidative phosphorylation system, 22 tRNAs, and 2 rRNAs. Yet, remarkably, over 90% of mitochondrial proteins are encoded by nuclear DNA and imported into the organelle, illustrating the deep interdependence between the two genomes.

Figure 2. Two Architectures of Life: The Linear Genome and Its Circular Counterpart. Gross structural organization of nuclear DNA and mitochondrial DNA. The nucleus houses the cell’s primary genome, arranged as linear chromosomes tightly packaged into chromatin around histone proteins. This diploid complement contains thousands of genes distributed across 23 chromosome pairs. In contrast, mitochondria carry their own compact genome: a small, circular DNA molecule present in hundreds to thousands of copies per cell. Unlike nuclear DNA, mitochondrial DNA lacks histones, resides within the mitochondrial matrix, and is inherited exclusively through the maternal line. Together, these two genomic architectures reflect the dual genetic systems that sustain human life.
Mitochondria and Cellular Energy
The primary role of mitochondria is to generate ATP, the universal energy currency of the cell, through oxidative phosphorylation. Because energy demands vary dramatically across tissues, the number of mitochondria per cell is not fixed. A single cell always contains one nucleus, but it may harbor hundreds to thousands of mitochondria, depending on its metabolic needs. For example, neurons, cardiomyocytes, and skeletal muscle fibers—cells with high energetic demands—contain some of the densest mitochondrial populations in the human body.
A Foundation for Understanding Health, Disease, and Neurodevelopment
This dual genome architecture—nuclear DNA and mitochondrial DNA—creates a complex genetic landscape that shapes cellular function, resilience, and vulnerability. As we move deeper into this series, we will explore how mitochondrial health, bioenergetic capacity, and genomic integrity influence not only systemic physiology but also neurodevelopment, including emerging links to autism spectrum disorder (ASD) where mitochondrial dysfunction is reported in up to 30–50% of individuals in some studies [5].
2. A Closer Look at Mitochondrial DNA
Cells across the body—especially in energy-hungry organs such as the brain, heart, skeletal muscle, kidneys, and liver—cannot function without a continuous supply of ATP (adenosine triphosphate). ATP acts as the cell’s universal energy currency, and its breakdown releases the chemical energy required for growth, repair, signaling, and survival.
How Mitochondria Make Energy
Inside each mitochondrion, ATP is generated through a precisely coordinated sequence of biochemical reactions. These reactions are carried out by specialized enzymes, many of which are encoded by mitochondrial DNA (mtDNA) itself. Although mtDNA is small—only 16.6 kb—it encodes 37 essential genes, including 13 proteins that form the core of the mitochondrial energy-producing machinery.
These reactions belong to the mitochondrial respiratory chain, also known as the electron transport chain (ETC). This chain is composed of five multiprotein complexes:
- Complex I (NADH:ubiquinone oxidoreductase)
- Complex II (succinate dehydrogenase)
- Complex III (cytochrome bc1 complex)
- Complex IV (cytochrome c oxidase)
- Complex V (ATP synthase)
Together, these complexes create the proton gradient that drives ATP synthesis. Each complex is built from dozens of proteins, and the instructions for making them are split between mtDNA and nuclear DNA. In fact, more than 1,000 nuclear genes are required for normal mitochondrial function, underscoring the deep interdependence between the two genomes.
Genetic Variation and Its Consequences
Every gene in our DNA—whether nuclear or mitochondrial—provides instructions for making a specific protein, the fundamental building block of cellular structure and function. Natural genetic variation is common and usually harmless; these benign differences are known as polymorphisms.
However, some variants alter the gene’s message in ways that disrupt protein function. When a genetic change interferes with normal growth, development, or physiology, it is classified as a pathogenic variant or mutation.
In the context of mitochondria, a pathogenic mtDNA variant can impair the production or function of enzymes in the respiratory chain. When these enzymes fail to operate efficiently, the mitochondria cannot generate sufficient ATP, leading to cellular energy deficiency. Because tissues like the brain and heart rely heavily on oxidative metabolism, they are particularly vulnerable to such deficits.
Why This Matters for Health and Neurodevelopment
Mitochondrial dysfunction—whether caused by mtDNA mutations, nuclear gene defects, or environmental stressors—can compromise ATP production, increase reactive oxygen species (ROS), and trigger metabolic stress. These disruptions are increasingly recognized in a range of conditions, including neurodevelopmental disorders. Notably, studies report evidence of mitochondrial abnormalities in 30–50% of individuals with autism spectrum disorder (ASD), highlighting the importance of understanding how mitochondrial genetics shapes brain development.
3. What It Means to Have a Mitochondrial DNA Variant
Every cell in the human body contains a population of mitochondria, ranging from only a few in lowenergy tissues to hundreds or even thousands in organs with high metabolic demands. Because each mitochondrion carries its own mitochondrial DNA (mtDNA), a person’s cells contain many copies of the mitochondrial genome.
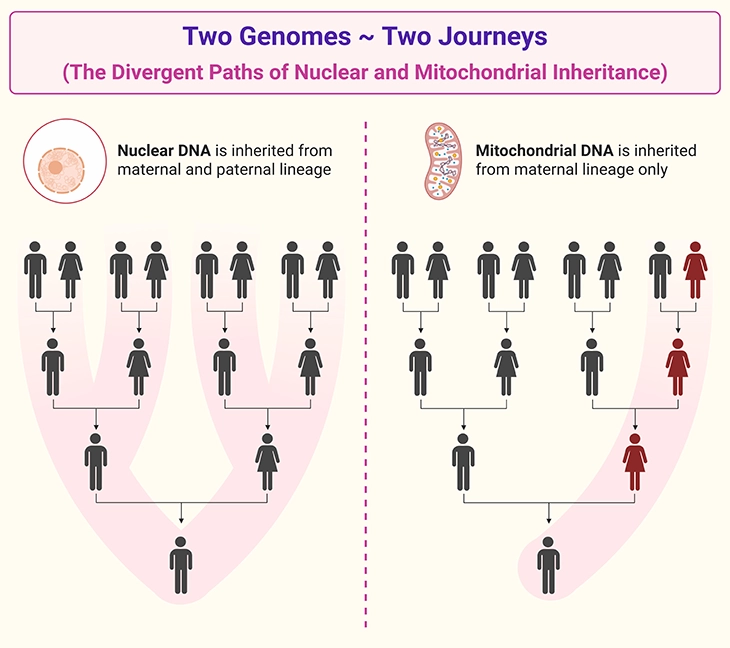
Figure 3. Two Genomes ~ Two Journeys: The Divergent Paths of Nuclear and Mitochondrial Inheritance. Contrasting patterns of inheritance for nuclear DNA and mitochondrial DNA. Nuclear DNA (left) is inherited from both parents, with each child receiving 50% of their nuclear genome from the mother and 50% from the father. Nuclear genes follow Mendelian inheritance patterns (autosomal dominant, autosomal recessive, Xlinked), and siblings may inherit different combinations of parental alleles. Mitochondrial DNA (right) is inherited exclusively from the mother, as mitochondria in the sperm are typically eliminated after fertilization. All children of an affected or carrier mother inherit her mitochondrial DNA, whereas fathers do not transmit mtDNA to their offspring. Because each egg contains a mixture of normal and mutant mitochondria (heteroplasmy), the proportion passed to each child varies, leading to variable expression and threshold-dependent symptoms across individuals and tissues.
Maternal Inheritance: The Mitochondrial Lineage
All of these mitochondria—and therefore all copies of mtDNA—originate from the mother’s egg cell at the moment of conception. While sperm do contain mitochondria, they are typically destroyed after fertilization, making paternal transmission of mtDNA exceedingly rare. Thus, mtDNA follows a maternal inheritance pattern, passing from mother to child through the egg [1-4].
This means that a pathogenic variant in a mitochondrial gene can be transmitted from a mother to all her children, though the severity and expression of the condition may vary widely (see Figure 1; 3; and 4).
When All Mitochondria Carry the Same Variant
An egg cell contains hundreds of thousands of mitochondria. If every mitochondrion in that egg carries a nonfunctional mtDNA variant, the resulting embryo may experience such profound energy failure that development cannot proceed, often leading to early embryonic loss. This reflects the essential role of mitochondrial ATP production in supporting rapid cell division and organ formation.
The Complexity of Dual Genomes
However, predicting outcomes is not always straightforward. Mitochondrial function depends on the interaction between mtDNA and nuclear DNA, which together encode more than 1,000 proteins required for mitochondrial structure, metabolism, and repair. Variants in either genome—or in both—can modify the impact of a mitochondrial mutation, sometimes softening or intensifying its effects.
Heteroplasmy: A Mixture of Healthy and Mutant Mitochondria
Most individuals with mtDNA variants do not have a uniform population of mitochondria. Instead, cells often contain a mixture:
- some mitochondria with a working copy of the gene
- others with a nonworking or partially working copy
This mixture is called heteroplasmy, and it is one of the most important concepts in mitochondrial genetics (see Figure 1) [1-4].
The proportion of mutant to normal mtDNA determines whether a cell can meet its energy needs. Even if some mitochondria function normally, the overall ATP output may fall below the threshold required for healthy tissue function, leading to a mitochondrial disorder.
When Variants Cause No Detectable Problem
On the other hand, if the proportion of mitochondria with a nonworking gene remains below a critical threshold—often around 60–80%, depending on the tissue—the cell may still produce enough ATP to function normally. In such cases, a person may carry a mitochondrial variant without showing symptoms, or symptoms may emerge only under metabolic stress, illness, or aging.
This threshold effect explains why mitochondrial conditions can vary dramatically within families, between siblings, and even between different tissues in the same individual (see Figure 1).
4. An Example of Mitochondrial (Maternal) Inheritance
Sometimes a mitochondrial DNA (mtDNA) variant arises for the first time in the mother’s egg or at the moment of fertilization. This type of change—called a de novo or spontaneous variant—creates a nonworking mitochondrial gene in the affected individual. When this happens, the person becomes the first in the family to show the condition, a pattern described as sporadic. If the affected individual is female, she can pass this mtDNA variant to her children through her egg cells.
Inherited Variants and the Maternal Line
More commonly, however, a mitochondrial variant is inherited from a mother whose cells—including her egg cells—contain a mixture of working and nonworking copies of the mitochondrial gene. This mixture reflects heteroplasmy, the coexistence of normal and mutant mtDNA within the same cell (see Figure 1; and 4) [1-4].
A typical family tree illustrating this pattern shows a grandmother (marked as A in Figure 4) who carries one or more nonworking mtDNA variants but remains clinically unaffected because she retains enough functional mitochondria to meet her cells’ energy needs.

Figure 4. Through the Maternal Line: A Quiet Mutation’s Journey Across Generations. Maternal transmission of a nonworking mitochondrial DNA (mtDNA) gene variant across generations. This pedigree illustrates how a grandmother (marked as A) carrying a mixture of working and nonworking mtDNA copies (heteroplasmy) can pass the variant to all her children, regardless of sex. Because mitochondria are inherited exclusively through the egg, only females transmit the variant to the next generation, while affected males (marked as B) do not pass it on. The proportion of nonworking mtDNA varies among offspring due to random segregation during egg formation, leading to differences in whether individuals are unaffected carriers, mildly affected, or clinically affected. Disease expression emerges only when the mutant load in critical tissues exceeds a functional threshold, explaining the variability in symptoms within the same family.
Why Not All Children Are Affected
Although the grandmother passes her mitochondria to all her children, not every child will necessarily develop the condition. This is due to the threshold effect—a fundamental principle of mitochondrial genetics.
During egg formation, mitochondria are randomly distributed into each developing egg. As a result, each egg may contain a different proportion of working and nonworking mitochondria, ranging from mostly normal to mostly abnormal. This randomness means:
- All of the grandmother’s children—sons and daughters—inherit some nonworking mitochondria.
- A child will only develop symptoms if: (a) the proportion of nonworking mtDNA exceeds a critical threshold, and (b) enough tissues in the body accumulate cells above this threshold to impair ATP production.
Thus, two of the grandmother’s children may remain unaffected because they inherited sufficient working copies to maintain normal mitochondrial function (see Figure 4).
Why Fathers Do Not Pass on mtDNA Conditions
In the example, the father (marked as B in Figure 4) has the mitochondrial condition, but his children will not inherit it, because mtDNA is almost exclusively passed through the mother’s egg. Even if a father is affected, his sperm mitochondria are typically eliminated after fertilization.
Why Daughters Can Still Pass It On
The grandmother’s daughters, however, can pass the variant to their children—even if they themselves show no symptoms. The risk is difficult to predict because it depends on:
- the proportion of nonworking mitochondria in the egg at conception,
- which tissues and organs in the developing child accumulate enough mutant mtDNA to cross the critical threshold, and
- the modifying influence of nuclear genes, which can either buffer or exacerbate mitochondrial dysfunction.
This interplay of heteroplasmy, threshold effects, and maternal inheritance explains why mitochondrial conditions can vary so widely within the same family—sometimes dramatically so (see Figure 1; 3; and 4) [1-4; 6].
TakeHome Messages
- Mitochondria carry their own DNA, inherited almost exclusively from the mother, forming a unique genetic lineage within every cell.
- Energy-hungry organs—the brain, heart, muscle, liver, and kidneys—depend profoundly on mitochondrial integrity and ATP production.
- Mitochondrial DNA variants can arise spontaneously or be maternally inherited, but their impact depends on the proportion of working vs. nonworking mitochondria.
- Heteroplasmy—the mixture of normal and mutant mtDNA—is central to why mitochondrial conditions vary so widely within families.
- A mitochondrial variant causes disease only when the mutant load crosses a critical threshold in enough tissues to impair energy production.
- Fathers may be affected by mitochondrial disease, but do not pass mtDNA to their children; only mothers transmit the mitochondrial genome.
- The interplay between mtDNA and nuclear DNA adds complexity, modifying severity, symptoms, and clinical outcomes.
- Mitochondrial dysfunction is not binary; it exists on a spectrum shaped by thresholds, tissue vulnerability, and metabolic demand.
- These principles form the foundation for understanding how mitochondrial health influences development, including emerging links to autism spectrum disorder.
(Cf. previous blogs entitled as: “Cellular Respiration: The Hidden Engine Driving Life’s Energy.”; “ATP: The Molecular Currency That Keeps Life Running.”)
Summary and Conclusions
Mitochondria, with their dual genomic heritage and central role in ATP production, form one of the most intricate and consequential systems in human biology. Their unique maternal inheritance, the phenomenon of heteroplasmy, and the threshold effect together create a genetic landscape where the same mitochondrial variant can manifest with striking variability — from silent carrier states to profound multisystem disease. This complexity is not merely academic; it shapes how we understand vulnerability, resilience, and clinical expression across the lifespan.
The principles explored in this series highlight several enduring truths. First, mitochondrial function is foundational to the health of organs with high metabolic demand — especially the brain, where energy availability governs synaptic development, neuronal signaling, and circuit maturation. Second, the interplay between mtDNA and nuclear DNA introduces layers of modulation that can either buffer or amplify the effects of mitochondrial variants. Third, the random segregation of mitochondria during egg formation and early embryogenesis creates a mosaic of cellular energy capacities, explaining why mitochondrial disorders often defy simple Mendelian expectations.
These insights carry profound implications for the neurodevelopmental, neuropsychiatric, and neurodegenerative communities. Mitochondrial dysfunction has been increasingly implicated in conditions such as autism spectrum disorder, ADHD, epilepsy, schizophrenia, bipolar disorder, Parkinson’s disease, and Alzheimer’s disease. In autism alone, abnormalities in mitochondrial enzymes, oxidative stress markers, and ATP production have been reported in 30–50% of individuals in some studies. Recognizing the principles of mitochondrial inheritance — especially heteroplasmy and threshold effects — is essential for interpreting these findings and for understanding why symptoms may vary so widely between individuals and across tissues.
Yet, despite remarkable progress, important gaps in knowledge remain. We still lack precise tools to predict how a given mtDNA variant will behave across different tissues, developmental stages, or environmental contexts. The mechanisms governing mitochondrial segregation during oogenesis and early embryonic development remain incompletely understood. The interactions between mtDNA variants and nuclearencoded mitochondrial proteins — and how these interactions shape disease risk — are only beginning to be mapped. Moreover, the field still seeks reliable biomarkers that can distinguish benign mitochondrial variation from pathogenic dysfunction, especially in neurodevelopmental conditions where metabolic signatures may be subtle or dynamic.
Looking ahead, future directions must include deeper integration of mitochondrial genetics with systems neuroscience, developmental biology, and clinical phenotyping. Advances in singlecell sequencing, livecell imaging, metabolomics, and computational modeling offer unprecedented opportunities to unravel how mitochondrial heterogeneity shapes brain development and behavior. Understanding how environmental stressors — inflammation, toxins, metabolic load — interact with mitochondrial vulnerability will be essential for designing targeted interventions. And as therapies evolve, from metabolic support to genespecific approaches, appreciating the nuances of mitochondrial inheritance will be critical for counseling families, designing clinical trials, and interpreting therapeutic outcomes.
In closing, mitochondria are not merely cellular powerhouses; they are biological storytellers, carrying a maternal legacy that influences every stage of human life. Their genetics, their fragility, and their adaptability shape the trajectory of health and disease in ways we are only beginning to appreciate. For the communities working at the frontiers of neurodevelopment, psychiatry, and neurodegeneration, understanding mitochondrial inheritance is not optional — it is essential. It provides a framework for interpreting variability, identifying risk, and envisioning new pathways toward prevention, diagnosis, and care.
MitoSwab™
Authoritative Mitochondrial Assessment
MitoSwab™ delivers a non-invasive, clinically validated analysis of mitochondrial function. Using a simple buccal swab, it precisely quantifies Electron Transport Chain components and Citrate Synthase activity—a definitive marker of mitochondrial content.
Clinical Correlation: 84% agreement with the gold-standard muscle biopsy.
For the initial investigation of mitochondrial dysfunction, MitoSwab™ is the proven, practical alternative to invasive procedures.
Choose the standard of simplicity. Choose MitoSwab™.

References
- Wallace DC. Mitochondrial genetic medicine. Nat Genet. 2018 Dec;50(12):1642-1649. doi: 10.1038/s41588-018-0264-z. Epub 2018 Oct 29. PMID: 30374071.
https://pubmed.ncbi.nlm.nih.gov/30374071/
(A sweeping, authoritative review on mitochondrial inheritance, heteroplasmy, threshold effects, and disease — the single most essential reference.) - Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM. Mitochondrial diseases. Nat Rev Dis Primers. 2016 Oct 20;2:16080. doi: 10.1038/nrdp.2016.80. PMID: 27775730.
https://pubmed.ncbi.nlm.nih.gov/27775730/
(A definitive, highimpact primer covering mitochondrial genetics, clinical variability, and maternal inheritance patterns.) - Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015 Sep;16(9):530-42. doi: 10.1038/nrg3966. PMID: 26281784.
https://pubmed.ncbi.nlm.nih.gov/26281784/
(The most comprehensive modern review on heteroplasmy, bottleneck effects, and tissuespecific thresholds.) - Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012 Dec;13(12):878-90. doi: 10.1038/nrg3275. PMID: 23154810; PMCID: PMC3959762.
https://pubmed.ncbi.nlm.nih.gov/23154810/
(A classic, deeply cited review on mtDNA mutations, inheritance, and clinical consequences — foundational for your article.) - Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012 Mar;17(3):290-314. doi: 10.1038/mp.2010.136. Epub 2011 Jan 25. PMID: 21263444; PMCID: PMC3285768.
https://pubmed.ncbi.nlm.nih.gov/21263444/
(The landmark metaanalysis linking mitochondrial abnormalities to ASD [30–50% prevalence in some cohorts].) - Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012 Mar 16;148(6):1145-59. doi: 10.1016/j.cell.2012.02.035. PMID: 22424226; PMCID: PMC5381524.
https://pubmed.ncbi.nlm.nih.gov/22424226/
(A beautifully written, highimpact synthesis of mitochondrial biology, disease mechanisms, and cellular energetics.)


